Native Methylation Meets Targeted Precision
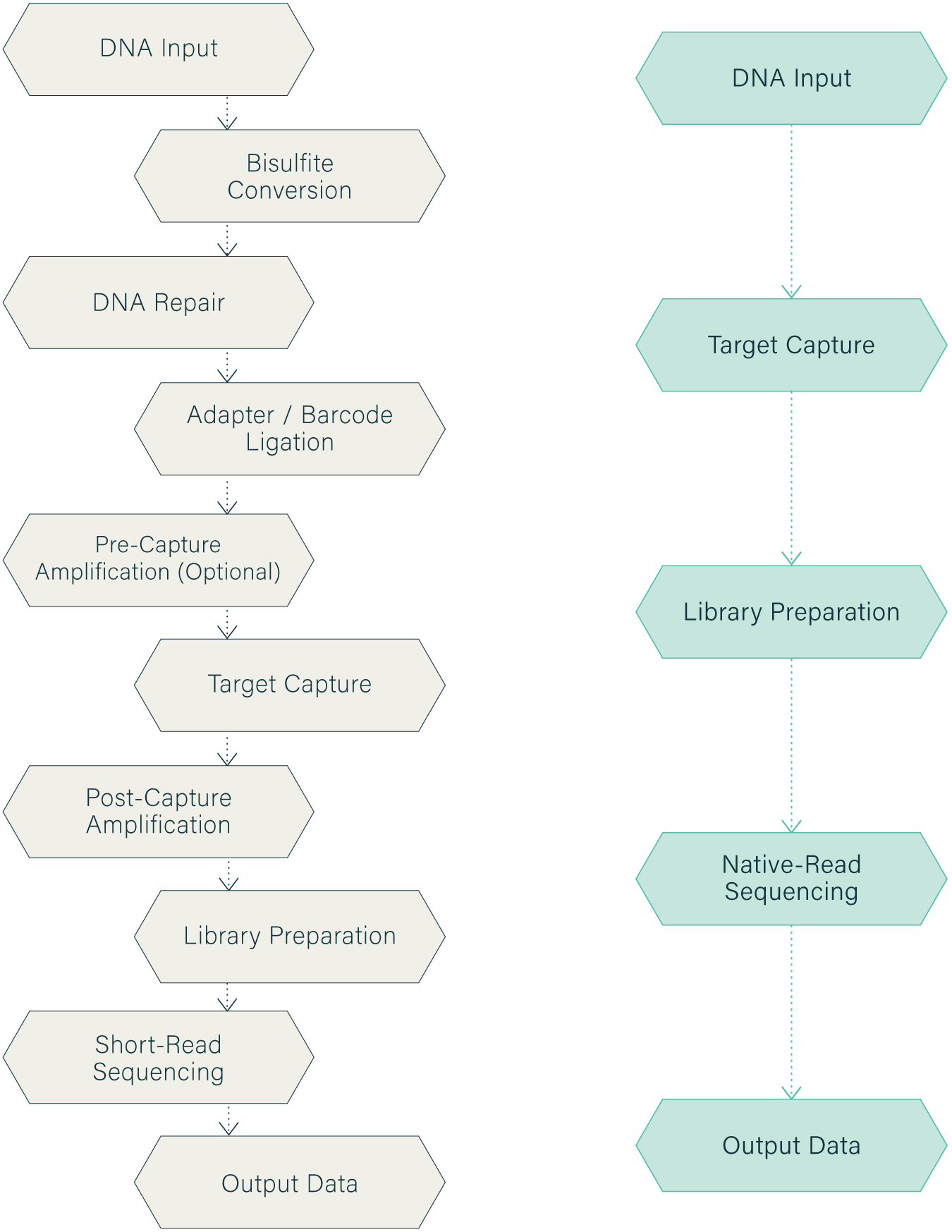
Wasatch BioLabs’ Direct Targeted Methylation Sequencing (dTMS) is a scalable, cost-effective platform for targeted long-read sequencing and methylation analysis, designed for applications where whole-genome context is unnecessary or cost-prohibitive.Wasatch BioLabs' Direct Targeted Methylation Sequencing (dTMS) provides a scalable, cost-effective solution for region-specific analysis, ideal for applications where whole-genome sequencing is inefficient or expensive. Combining hybridization-based enrichment with nanopore sequencing, dTMS overcomes the limitations of traditional methods, including short-reads and synthetic DNA that erase native modifications.
By combining proprietary library prep protcols, modified Agilent SureSelect hybridization-based enrichment, and native, long-read Oxford Nanopore sequencing, dTMS overcomes the limitations of short-read and PCR-based approaches that can obscure structural variation and eliminate native epigenetic modifications. The result is high-depth, multiplexed interrogation of defined genomic regions while preserving methylation and long-range haplotypic context.